1.Tyrosine(酪胺酸)
|
A.相關疾病(a.b.c.疾病請對應下方圖片的a.b.c.之處)
a. 酪胺酸血症(Tyrosinemia):別名「TypeⅡTyrosinemia」
I.因為缺乏Tyrosine transaminase,無法合成p-Hydroxyphenylpyruvate
II.酪胺酸血症的臨床表現主要在肝臟
III.血漿中tyrosine、phenylalanine濃度升高。患者會發生皮膚和眼睛的損害和心智遲滯
b.嬰兒酪胺酸血症(Neonatal Tyrosinemia)
I.缺乏p-hydroxyphenylpyruvate hydroxylase。血漿中tyrosine、phenylalanine升高
II. 酪胺酸血症的臨床表現主要在肝臟及腎臟。患者會發生肝功能異常、黃疸,以及慢性腎衰竭
III.以往的治療效果不太好,不過最近已經有一些新的藥物在使用
註:以上這2個的治療原則為吃low protein diet
c.黑尿症(Alkaptonuria)
I.黑尿病是一種罕見的遺傳病,患者的尿液會在排出後幾小時變成黑色。暫時仍沒有方法可以治療
II.患者的身體缺少了Homogentisate oxidase,不能正常的代謝酪氨酸,使得酪氨酸的代謝產物Homogentisate(尿黑酸)積聚體內,並排到尿液裡,而尿液在接觸空氣後變成黑色。
III.酪胺酸被polyphenyl oxidase氧化後,與結締組織之大分子會發生聚合而著色,使黑尿症末期發生褐黃病(Ochronosis)及關節炎(Arthritis)
IV.尿黑酸在耳朵的軟骨積聚,令耳朵顏色變深,在眼睛積聚,令鞏膜和角膜出現黑斑
 |
2.Phenylalanine(苯丙胺酸)
| |
A.苯酮尿症(Phenylketonuria,PKU)
a.1934 年挪威的Dr.Folling 在家族性智障病患的尿液中發現有特殊陳腐味道,後來才知道該物質是苯丙酮酸(phenylpyruvic acid),這和尿中有糖的糖尿病是無關的。
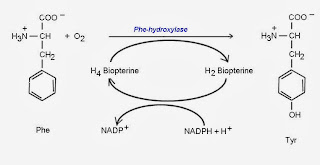
b.苯酮尿症是一種體染色體隱性遺傳疾病,主要是由於體內苯丙胺酸 (phenylalanine;Phe)羥化(hydroxylation)成酪胺酸(tyrosine;Tyr)的代謝途徑機障所引起的先天代謝異常疾病。
| |
正常情況
Phe hydroxylase正常作用將Phe轉變成Tyr
|
苯酮尿症
缺乏Phe hydroxylase,直接進行轉胺作用,生成苯丙酮酸
|
 資料來源:campbell |  資料來源:生物資訊 |
3.Valine、Isoleucine、Leucine(缬氨酸、異白胺酸、白胺酸)
| ||||||||||||
A. Valine, isoleucine和leucine皆為支鏈胺基酸(branched-chain amino acid,簡稱BCAA),皆會經由轉胺作用、氧化性脫羧作用(*α-酮基酸脫氫酶),最後經脫氫作用等氧化路徑代謝掉。
B.代謝路徑:
A.Maple syrup urine disease (MSUD;楓糖尿症)
a.新生兒由於先天遺傳缺陷,粒線體中缺乏「支鏈酮酸去氫酶」 (Branched-chain α-keto acid dehydrogenase;BCKD),導致leucine、isoleucine及valine等支鏈胺基酸無法進行脫羧作用以繼續代謝反應,而使酮酸在體內累積
註:「支鏈酮酸去氫酶」就是α-酮基酸脫氫酶(α-keto acid dehydrogenase)
b.尿中有一種特殊的楓糖漿氣味故稱楓糖尿症
c. MSUD 會有生理及心理發育遲緩,患嬰生長不良,有陣發性腦病變及酸中毒等症狀
d. 發生率:1/120,000 ~ 1/150,000
e.治療方式:終身需營養治療
I.早期使用低蛋白或設計過之飲食(不含支鏈胺基酸的飲食)
|

胺基酸與草醋酸形成(amino acid and Oxaloacetate formation)
胺基酸與α-酮戊二酸形成(amino acid and α-ketoglutarate formation)
胺基酸與丙酮酸形成(amino acid and pyruvate formation)
胺基酸與乙醯輔酶A(amino acid and acetyl-CoA formation)
▼有機物代謝(Organic matter metabolism)
顯示/隱藏(show/hide)
標籤:
代謝(metabolism)
0 意見:
張貼留言