因為這些疾病的每一種都比較罕見,作為一大類疾病,其在存活出生嬰兒中的發病率約為 1: 7,700﹔因此醫師在臨床實踐中將它們看作一個疾病類型。
大多數溶小體儲積症 (LSD) 的臨床嚴重程度都具有多樣性,呈現出連續系譜特性。 它們在本質上都具有進行性(不斷惡化)特徵,並可能導致身體全身性的不可反逆損傷,同時具嚴重衰退性,嚴重表現型甚至具有致命性。 因此,早期鑑別和診斷極為重要。
溶小體儲積症 (LSD) 需採用多種學科的組合方法治療與照護,某些溶小體儲積症有多種針對疾病的治療選擇及醫療控制措施。
資料來源:mps1
一、溶小體儲積症(Lysosomal storage disease)症狀
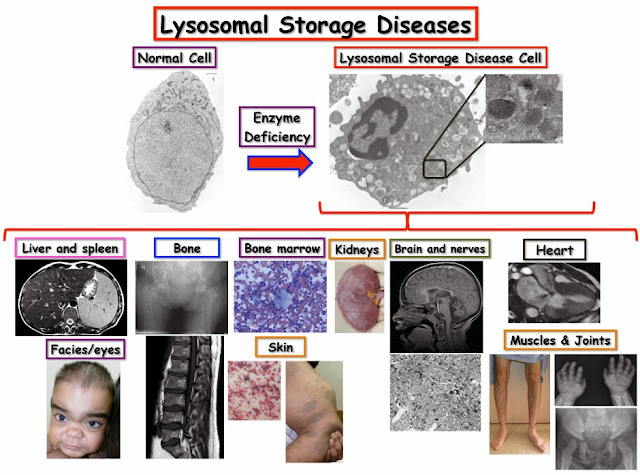
二、溶小體儲積症(Lysosomal storage disease)治療

資料來源:約翰霍普金斯醫療機構
▼細胞(Cell)
顯示/隱藏(show/hide)
標籤:
細胞(cell)
0 意見:
張貼留言